

Речевая аналитика — обеспечивает конвертацию разговоров в текстовый формат, что открывает дополнительные возможности для

На примере сервиса Ucaller, предоставляющего такой сервис, можно увидеть, что доставлять коды можно двумя

Profit Server уже почти 10 лет работает в сфере предоставления услуг по аренде выделенных

Ищете хостинг или виртуальный сервер с комфортными условиями по оплате и использованию? Такие решения

Перечень операций в рамках настройки 1С и стоимость услуг зависит от специфики компании, внедряемого/настраиваемого

При заказе VPS/VDS от CloudVPS можно воспользоваться готовыми дистрибутивами для быстрого старта.

Эквайринг – это процесс обработки платежей с использованием банковских карт, который стал неотъемлемой частью

С развитием технологий и расширением возможностей онлайн-связи, это становится все более привлекательным и востребованным

JUSTHOST — провайдер услуг, у которого есть все необходимое для уверенного присутствия в интернете.

Согласно статистике ежегодно российские маркетплейсы растут x2. Это касается, как заработка самих площадок, так
Regfon, как комплексное решение, включает услуги по продаже/аренде виртуальных номеров, ВАТС и оборудования для

Виртуальный номер телефона США (1+) – это удобный и эффективный способ общения и ведения
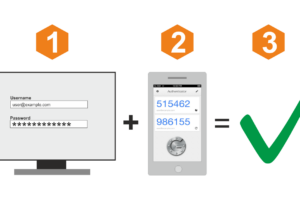
Ниже — обзор популярных сервисов авторизации по звонку, с помощью которых вы сможете быстро

Теплые полы – это современная система обогрева, которая обеспечивает равномерное распределение тепла по поверхности

Зимой уборка крыш от снега альпинистами очень необходима для безопасности и комфорта жителей города.

Системный интегратор информационно-коммуникационных технологий (ИКТ) является ключевым звеном в цепочке создания эффективных и гармонично

С течением времени технологии стремительно развиваются, и то, что казалось современным и актуальным несколько

Различные типы ковров требуют разных подходов к стирке. Например, натуральные шерстяные ковры требуют более
Полиграфия является важной составляющей современного мира коммуникаций.

Со временем ванные комнаты могут подвергаться износу и старению. Однако, перед тем как считать,

Венки можно встретить на многих захоронениях. Выбирают их в дань памяти ушедшему, показывая свое

Выбор виртуального номера телефона может быть сложной задачей, но существует несколько факторов, которые следует

Веб-шрифты – это особые шрифты, которые загружаются и отображаются на веб-страницах, позволяя веб-дизайнерам использовать

Восстановление секторов жесткого диска - это процесс восстановления поврежденных или неисправных секторов на жестком
В современном мире, где коммуникация играет ключевую роль в развитии бизнеса, виртуальные номера телефонов
Регистрация медицинских изделий является важной процедурой, предшествующей их введению на рынок и использованию в

Смартфоны стали неотъемлемой частью нашей повседневной жизни. Мы используем их для общения, развлечений и

Производство холодильного оборудования – пошаговая инструкция по организации бизнеса. Анализ рынка + регистрация ИП

Интерьерная печать – это уникальный и эффективный способ преобразить обычное пространство в неповторимое и